High-throughput sequencing technology generates vast amounts of short DNA sequence data, presenting both significant challenges and exciting opportunities in the field of bioinformatics. This article explores the key research areas such as data compression techniques, metagenomic sequence assembly, and advanced algorithms for analyzing complex genomic datasets. It also discusses future trends in handling short-read DNA sequences within high-throughput environments.
High-Throughput Sequencing AnalysisHigh-throughput sequencing, also known as next-generation or deep sequencing, allows the parallel analysis of millions to billions of DNA molecules simultaneously. This powerful technique enables detailed studies of transcriptomes and genomes, offering insights into gene expression, genetic variation, and functional genomics. Commonly referred to as NGS, it is a cornerstone of modern biological research.
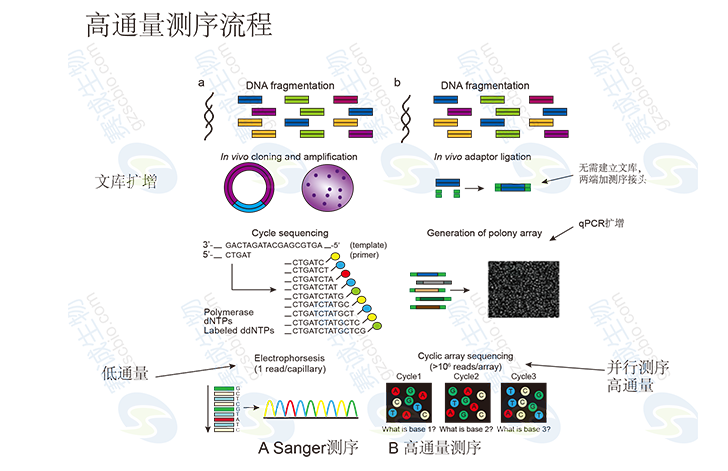
Figure 1: High-throughput sequencing process
Applications of High-Throughput SequencingHigh-throughput sequencing has a wide range of applications across various fields, including:
- 1. DNA sequencing: de novo genome sequencing, resequencing, metagenomic sequencing, and exome capture sequencing.
- 2. RNA sequencing: transcriptome profiling, small RNA analysis, and digital gene expression (DGE) studies.
- 3. Epigenetic studies: ChIP-Seq for protein-DNA interactions and DNA methylation analysis.
Genomic sequencing involves the high-throughput sequencing of an organism’s entire genome. It can be divided into two main approaches: de novo sequencing, used when no reference genome is available, and resequencing, which compares individual genomes to a known reference. These methods help identify genetic variations and support evolutionary studies.
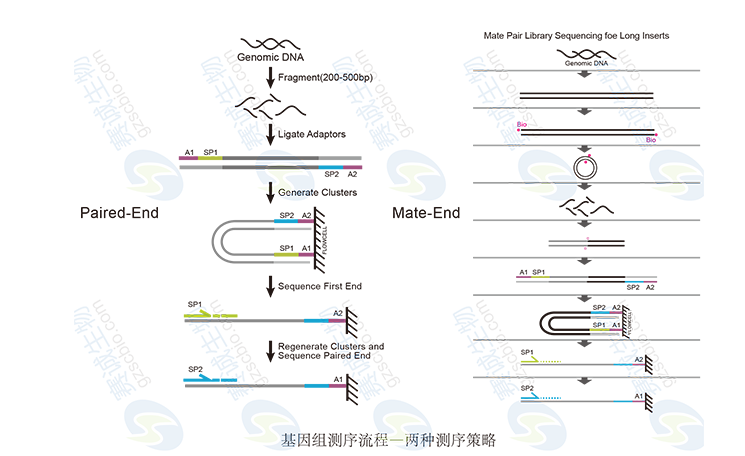
Figure 2: Genomic sequencing strategy
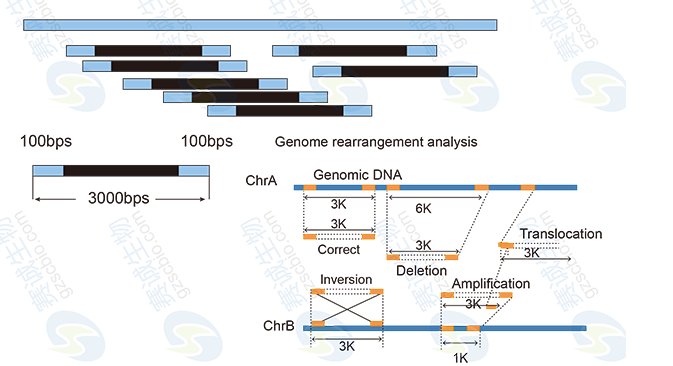
Figure 3: Paired-end principle
The paired-end method involves fragmenting the genome, ligating adapters, and sequencing both ends of the fragments. In contrast, mate-end sequencing involves more complex steps, including biotin labeling, cyclization, and enrichment before sequencing. These techniques enhance accuracy and coverage in genomic studies.
Bioinformatics plays a crucial role in genomic analysis, covering tasks such as data preprocessing, genome assembly, annotation, and comparative genomics. These steps are essential for interpreting the vast amount of sequencing data generated.
Metagenomic SequencingMetagenomic sequencing enables the study of microbial communities in diverse environments like soil, water, and the human gut. Unlike traditional culture-based methods, it allows direct analysis of all microorganisms present without prior isolation. Two common approaches are whole-genome metagenomics and 16S/18S rRNA sequencing, each offering unique insights into community structure and function.
Whole-genome metagenomics provides comprehensive insights into species diversity, functional potential, and metabolic pathways. On the other hand, 16S/18S rRNA sequencing focuses on identifying and quantifying microbial taxa, making it ideal for ecological and clinical studies.
Human Exome Capture SequencingExome sequencing targets the coding regions of the genome, where most disease-related mutations occur. Compared to whole-genome sequencing, it offers deeper coverage, higher accuracy, and lower cost, making it a popular choice for clinical and research applications.
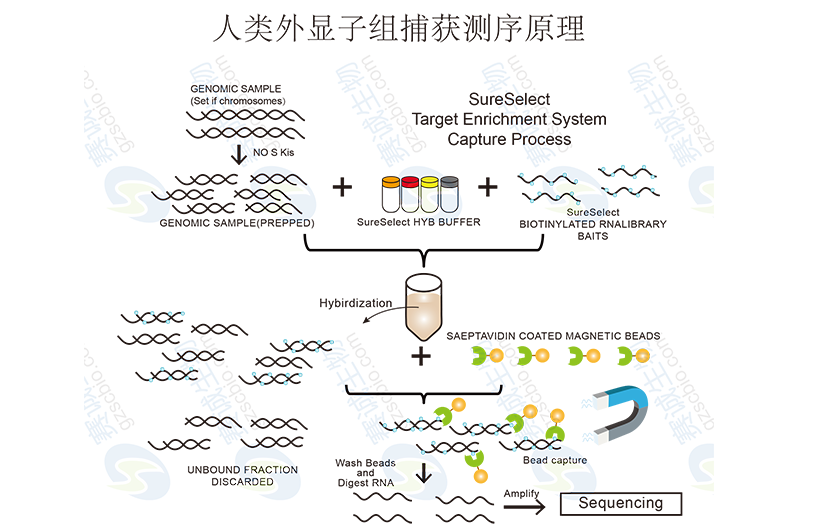
Figure 4: Human exome capture sequencing principle
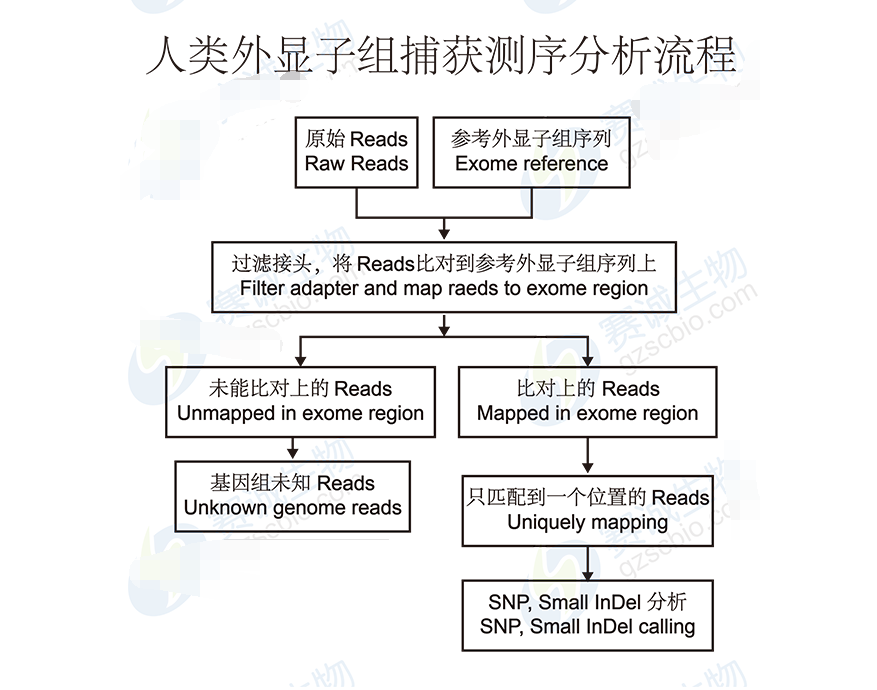
Figure 5: Human exome capture sequencing analysis process
Exon capture involves hybridizing DNA to specific probes that target exonic regions, followed by sequencing. Popular platforms include Roche NimbleGen and Agilent SureSelect, which offer high specificity and efficiency in capturing exonic sequences.
Transcriptome SequencingTranscriptome sequencing captures all RNA molecules expressed in a cell under specific conditions, including mRNAs and non-coding RNAs. It provides valuable insights into gene expression patterns, alternative splicing, and novel transcripts, supporting functional genomics research.
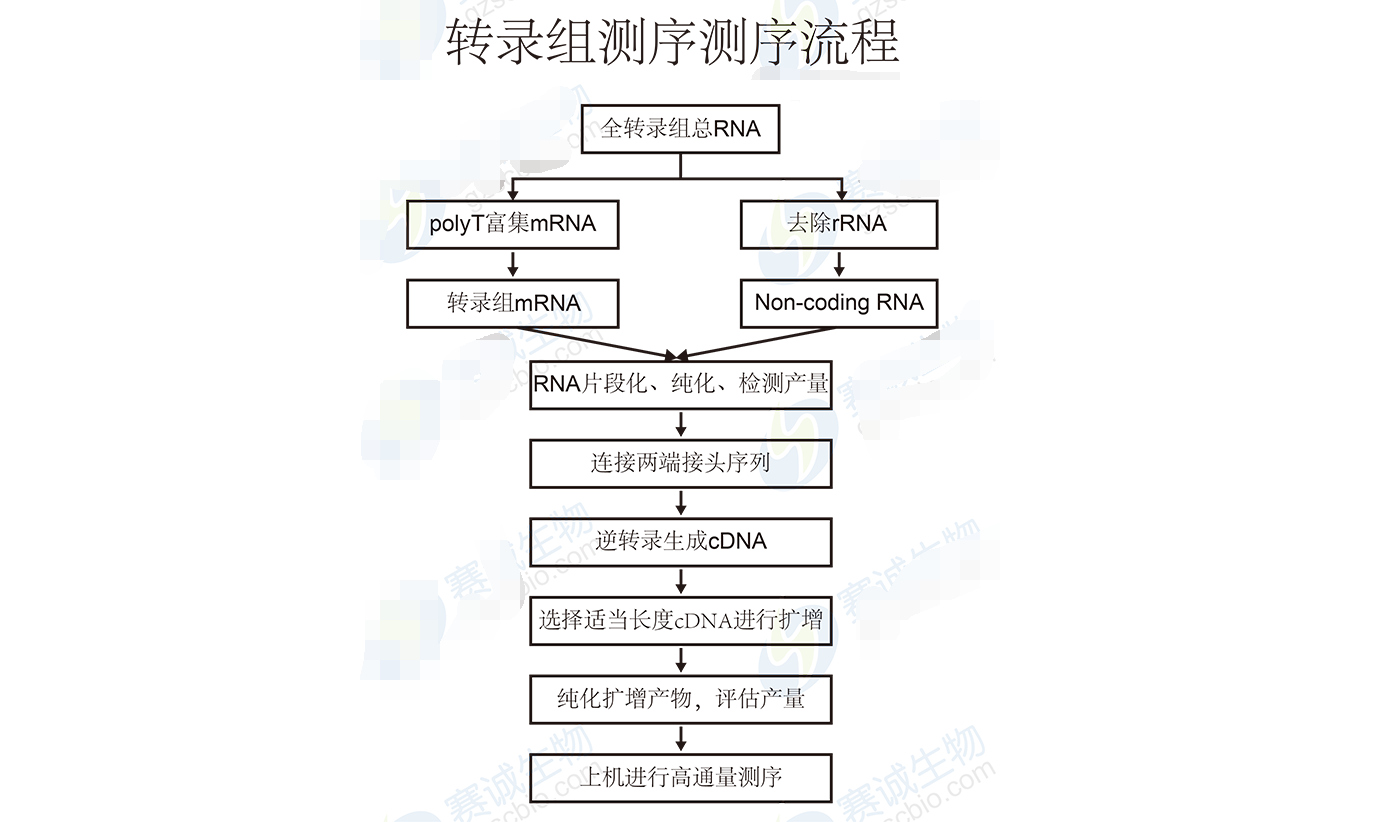
Figure 6: Transcriptome sequencing process
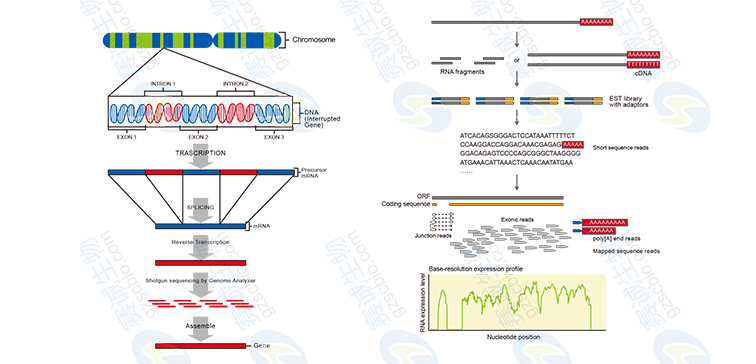
Figure 7: Transcriptome sequencing with and without a reference
Transcriptome analysis includes data quality assessment, assembly of transcripts, functional annotation, and differential expression studies. These analyses help uncover regulatory mechanisms and biological functions of genes.
Digital Gene Expression ProfilingDigital Gene Expression (DGE) is a cost-effective approach for studying gene expression by sequencing short tags from mRNA molecules. It provides accurate quantification of gene expression levels and is widely used in functional genomics and biomedical research.
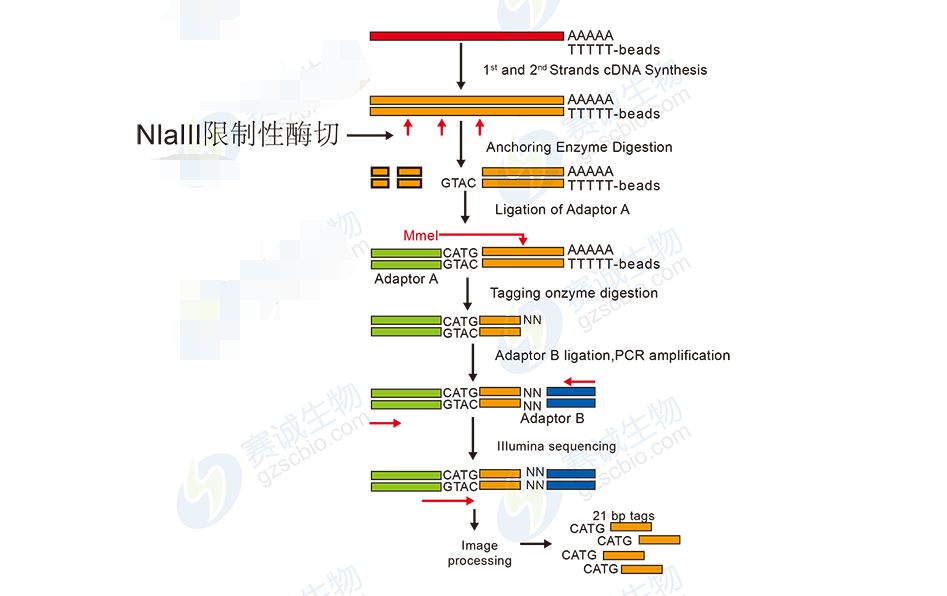
Figure 8: DGE sequencing workflow
DGE analysis includes base calling, data cleaning, tag counting, alignment, and differential expression analysis. It helps identify gene expression changes under different experimental conditions and contributes to understanding cellular processes.
Small RNA SequencingSmall RNAs, typically 21–31 nucleotides long, play critical roles in gene regulation through mechanisms such as mRNA degradation and transcriptional silencing. They include miRNAs, siRNAs, and piRNAs, each with distinct functions in development and disease.
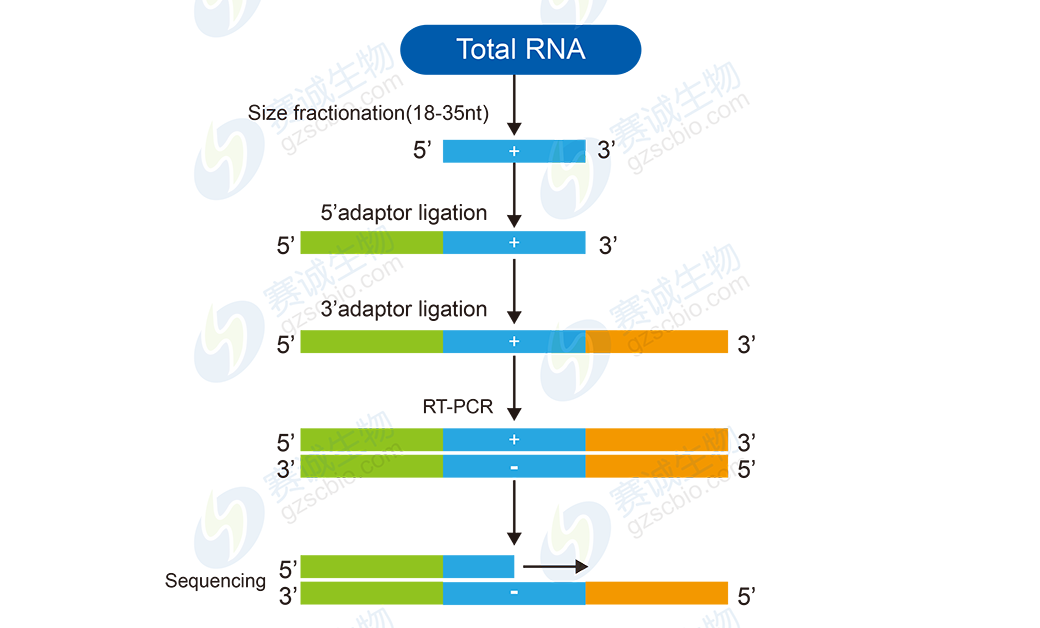
Figure 9: Small RNA sequencing workflow
Small RNA analysis involves basic steps like data filtering and length distribution analysis, as well as advanced features such as miRNA prediction and differential expression studies. These analyses help reveal the regulatory roles of small RNAs in biological systems.
ChIP-SeqChIP-Seq is a powerful technique for studying protein-DNA interactions, particularly for mapping transcription factor binding sites and histone modifications. It combines chromatin immunoprecipitation with high-throughput sequencing to generate genome-wide interaction maps.
ChIP-Seq AnalysisChIP-Seq analysis includes aligning sequencing reads to a reference genome, identifying enriched regions (peaks), and performing functional enrichment analysis. These steps help understand the biological significance of protein-DNA interactions and their impact on gene regulation.
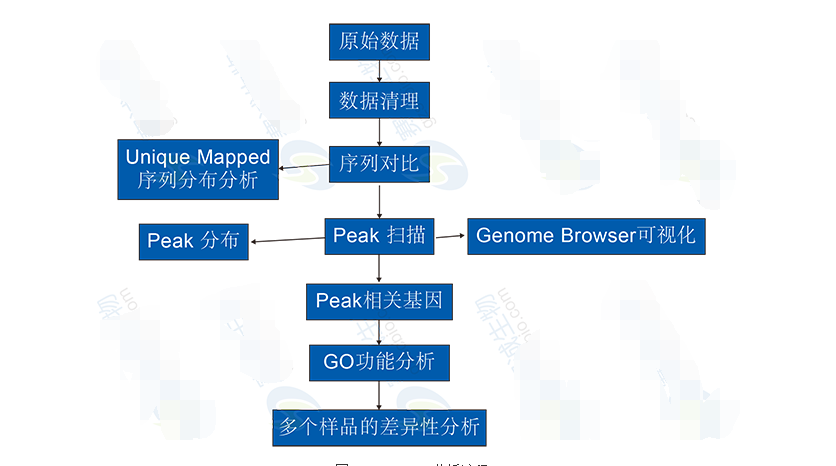
Figure 10: ChIP-Seq analysis process
DNA Methylation SequencingDNA methylation is a key epigenetic modification involved in gene regulation and cellular differentiation. Two major techniques for methylation analysis are MeDIP-Seq and Bisulfite-Seq, each offering unique advantages in detecting methylated regions across the genome.
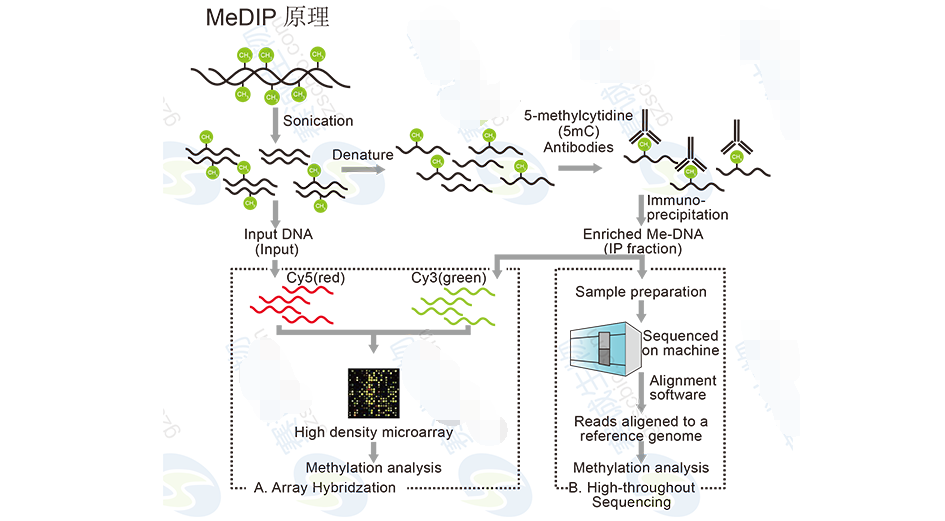
Figure 11: MeDIP principle
MeDIP-Seq AnalysisMeDIP-Seq involves immunoprecipitating methylated DNA fragments and sequencing them to identify regions of methylation. The analysis includes alignment, peak detection, and functional annotation, helping to uncover the role of methylation in gene regulation and disease.
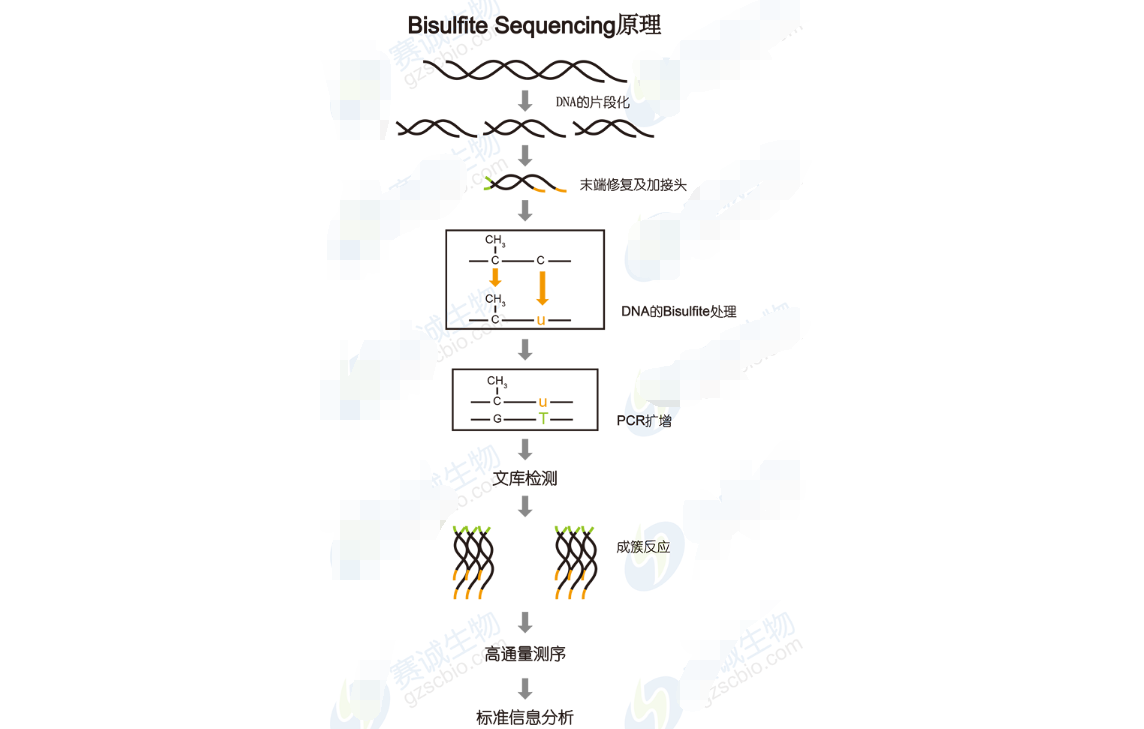
Figure 12: Bisulfite sequencing principle
Bisulfite Sequencing AnalysisBisulfite sequencing converts unmethylated cytosines to uracils, allowing precise detection of methylation patterns at single-base resolution. This method is widely used for studying epigenetic changes in development, disease, and environmental responses.
Analysis includes alignment, methylation level calculation, and genome-wide methylation pattern visualization. These insights contribute to understanding the dynamic nature of epigenetic regulation and its impact on cellular function.
2.54Mm Box Header,Smt Double Rows 2.54Mm Pitch Mainboard Connector,Box Header Circuit Board Connector,Box Header Mother Board Connections Parts
Dongguan City Yuanyue Electronics Co.Ltd , https://www.yyeconn.com