High-throughput sequencing technology generates short DNA sequence reads in massive volumes, presenting both significant challenges and opportunities in the era of big data. This review explores the key research areas, including data compression techniques, metagenomic sequence assembly, and advanced algorithms for analyzing complex genomic datasets. Furthermore, it provides a comprehensive outlook on the future development of short-read DNA sequencing under high-throughput platforms.
High-Throughput Sequencing AnalysisHigh-throughput sequencing, also known as next-generation sequencing (NGS) or deep sequencing, allows the parallel sequencing of millions to billions of DNA molecules simultaneously. This powerful technology enables detailed and comprehensive analysis of transcriptomes and genomes, offering insights into gene expression, genetic variation, and functional genomics. It is widely used across various biological disciplines due to its speed, accuracy, and cost-effectiveness.
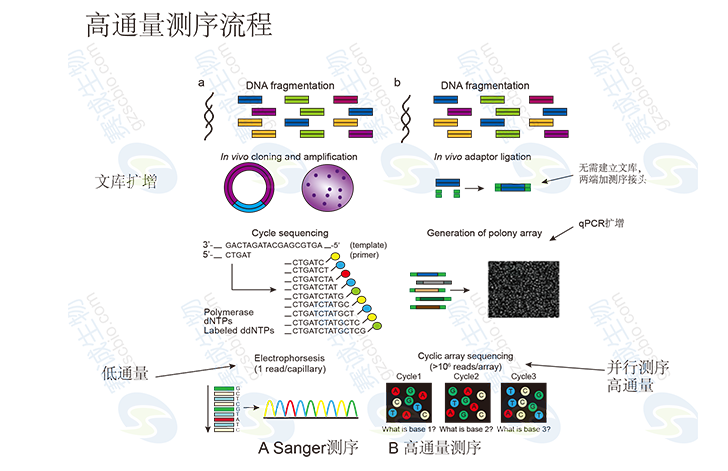
Figure 1: High-throughput sequencing process
Applications of High-Throughput SequencingHigh-throughput sequencing has broad applications in life sciences, including:
- DNA sequencing: Whole genome de novo sequencing, resequencing, metagenomic sequencing, and exome capture sequencing.
- RNA sequencing: Transcriptome profiling, small RNA detection, and digital gene expression (DGE) analysis.
- Epigenome studies: ChIP-Seq for protein-DNA interactions and DNA methylation sequencing for epigenetic regulation.
Genomic sequencing involves the high-throughput sequencing of an organism’s entire genome. It can be classified into two main types: de novo sequencing, which is used when no reference genome is available, and resequencing, which compares individual genomes to a known reference. The process includes data processing, genome assembly, annotation, and functional analysis, providing critical insights into genetic structure and function.
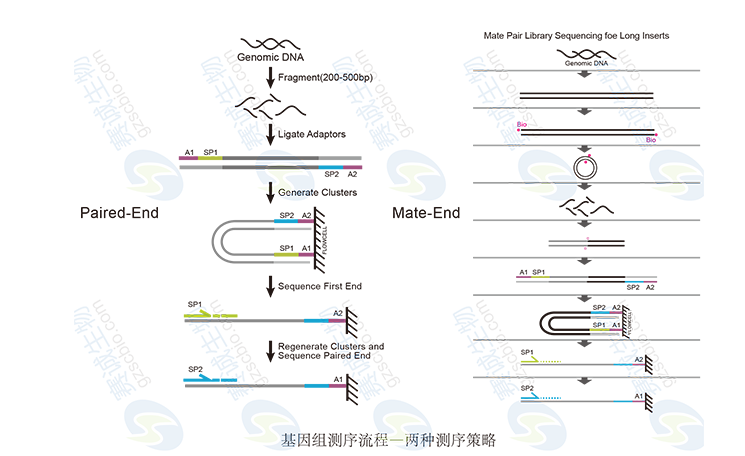
Figure 2: Genomic sequencing strategy
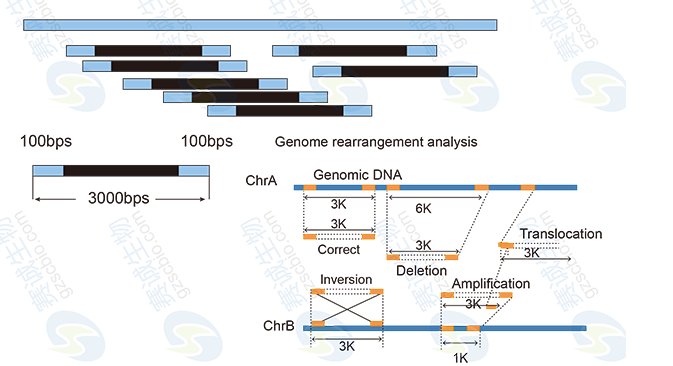
Figure 3: Paired-end principle
The paired-end method involves fragmenting the genome, attaching adaptors, and sequencing both ends of the fragments. Mate-pair sequencing follows a similar approach but with longer insert sizes, allowing for more accurate genome assembly and structural variant detection.
Genomic sequencing analysis typically involves several steps: data preprocessing, genome assembly, annotation, functional classification, and comparative genomics. These processes help researchers identify genetic variations, understand evolutionary relationships, and uncover functional elements within the genome.
Metagenome SequencingMetagenome sequencing refers to the sequencing of all microbial DNA present in a given environmental sample, such as soil, water, or the human gut. This technique enables the identification of microbial species, their abundance, and their functional roles within the community. Unlike traditional methods that require culturing, metagenomic sequencing can analyze unculturable microorganisms directly from environmental samples.
Two common approaches include whole-genome metagenomic sequencing, which captures the full genetic diversity of the microbial community, and 16S/18S rRNA sequencing, which focuses on taxonomic classification using conserved ribosomal RNA regions.
Metagenomic data analysis involves tasks such as sequence assembly, species classification, functional annotation, and comparative studies between different samples. These analyses provide valuable insights into microbial diversity, ecological functions, and potential biotechnological applications.
Human Exome Capture SequencingExome sequencing targets the protein-coding regions of the genome, known as exons, which make up only about 1–2% of the human genome. This approach offers deeper coverage, higher accuracy, and greater cost-efficiency compared to whole-genome sequencing. It is particularly useful for identifying mutations associated with genetic disorders and personalized medicine.
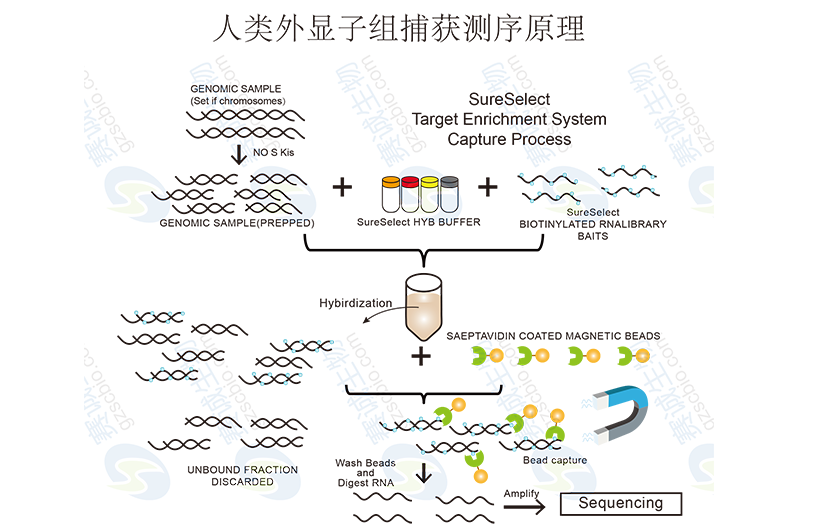
Figure 4: Principle of human exome capture sequencing
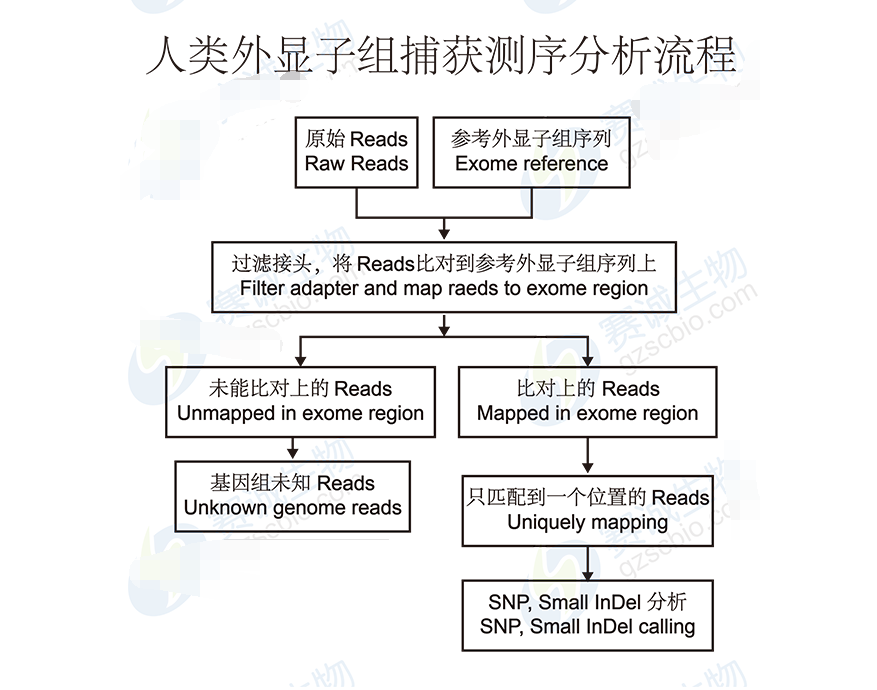
Figure 5: Human exome capture sequencing analysis process
Exon capture technologies, such as NimbleGen and Agilent SureSelect, are widely used to enrich the exonic regions before sequencing. This targeted approach ensures efficient use of resources and improves the detection of clinically relevant variants.
Transcriptome SequencingTranscriptome sequencing, or RNA-seq, provides a comprehensive view of the RNA molecules expressed in a cell or tissue at a specific time. It includes both coding and non-coding RNAs, enabling the study of gene expression, alternative splicing, and novel transcripts.
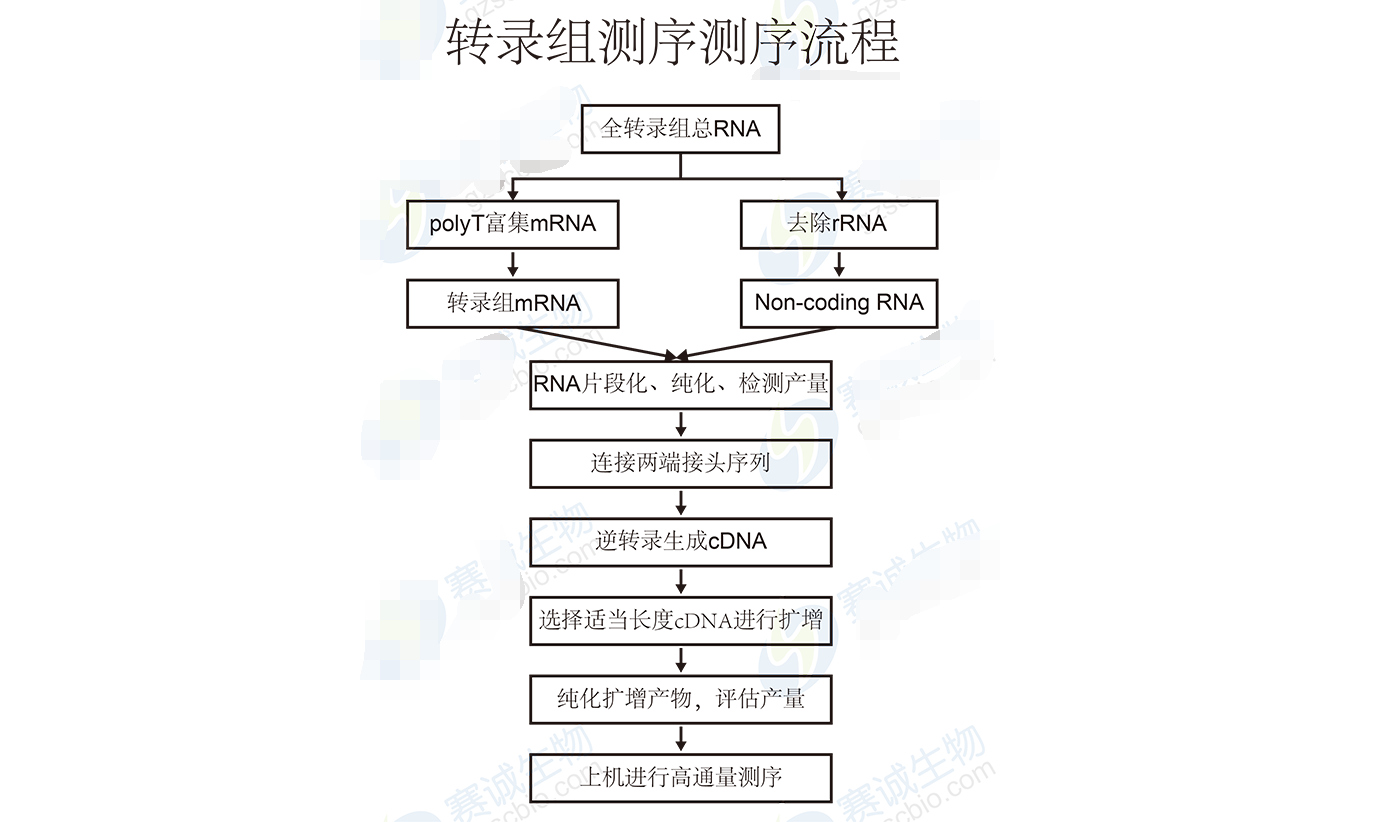
Figure 6: Transcriptome sequencing process
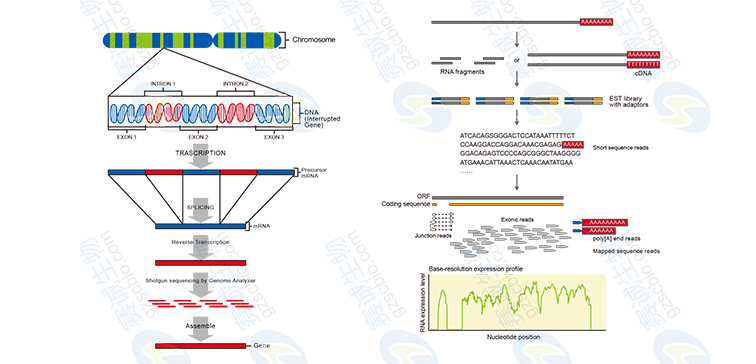
Figure 7: Reference-based and reference-free transcriptome sequencing
Transcriptome analysis involves multiple stages, including quality control, alignment, quantification, and differential expression analysis. With or without a reference genome, these analyses help researchers understand gene regulation, identify biomarkers, and explore disease mechanisms.
Digital Gene Expression ProfilingDigital gene expression (DGE) is a high-throughput sequencing method that captures short mRNA tags to profile gene expression levels. It is cost-effective and efficient for studying gene expression under various experimental conditions. DGE is widely applied in functional genomics and medical research.
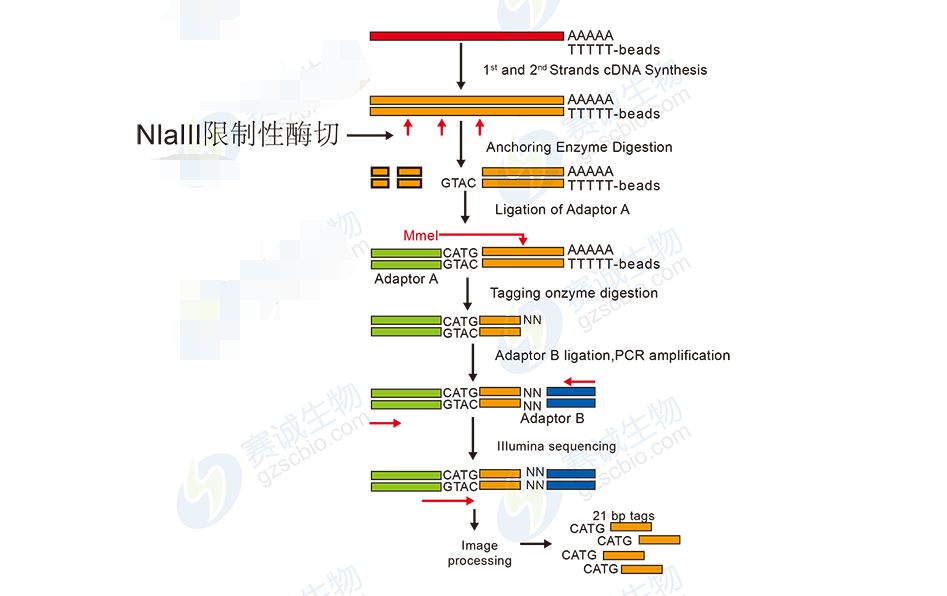
Figure 8: DGE sequencing workflow
DGE analysis includes steps such as image processing, base calling, filtering, tag counting, and alignment to reference sequences. It also involves differential expression analysis, clustering, and enrichment studies to identify key genes and pathways involved in biological processes.
Small RNA SequencingSmall RNAs, such as miRNAs, siRNAs, and piRNAs, play crucial roles in gene regulation at the transcriptional and post-transcriptional levels. Small RNA sequencing allows the identification and quantification of these regulatory molecules, revealing their functions in development, stress response, and disease.
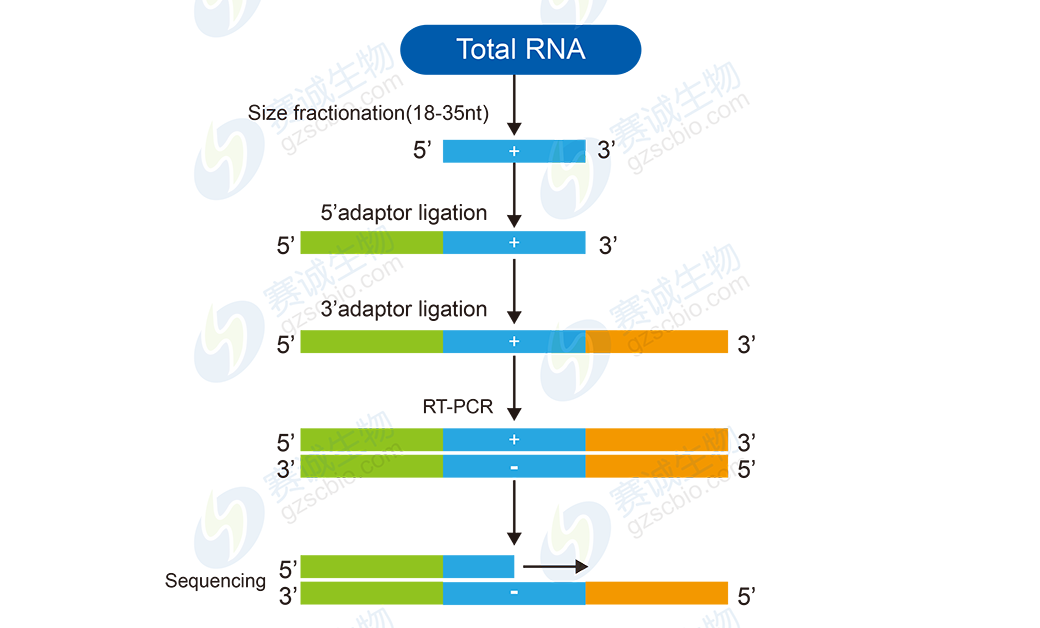
Figure 9: Small RNA sequencing workflow
Small RNA analysis typically includes basic steps like data preprocessing and alignment, followed by advanced tasks such as classification, novel miRNA prediction, and differential expression analysis. These insights help researchers understand the regulatory networks governed by small RNAs.
ChIP-SeqChIP-Seq (Chromatin Immunoprecipitation followed by sequencing) is a powerful technique used to study protein-DNA interactions, such as those involving transcription factors and histone modifications. It enables genome-wide mapping of binding sites and helps reveal the regulatory landscape of the genome.
ChIP-Seq Analysis Includes:1. Alignment of sequencing reads to a reference genome.
2. Genome-wide distribution of reads and peak detection.
3. Functional annotation of peaks and gene enrichment analysis.
4. Comparative analysis between multiple samples to identify differentially bound regions.
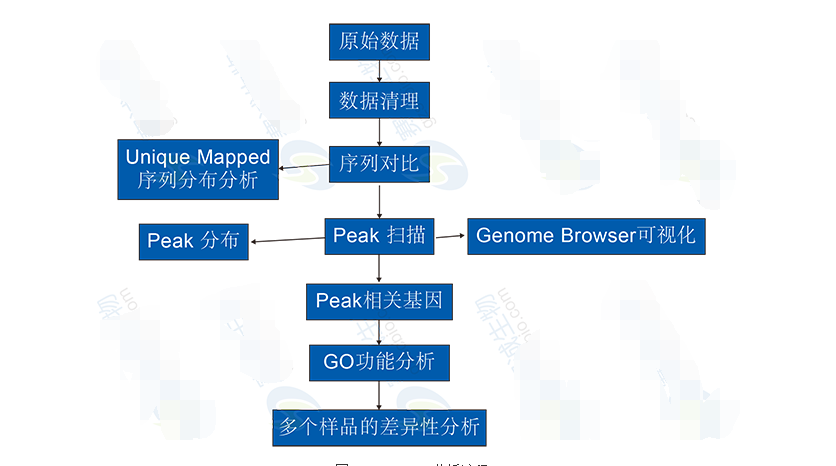
Figure 10: ChIP-Seq analysis process
DNA Methylation SequencingDNA methylation is a key epigenetic modification that influences gene expression and plays a role in development and disease. Two major sequencing methods for studying DNA methylation are MeDIP-Seq and Bisulfite Sequencing. Both allow researchers to map methylated regions across the genome and investigate their functional implications.
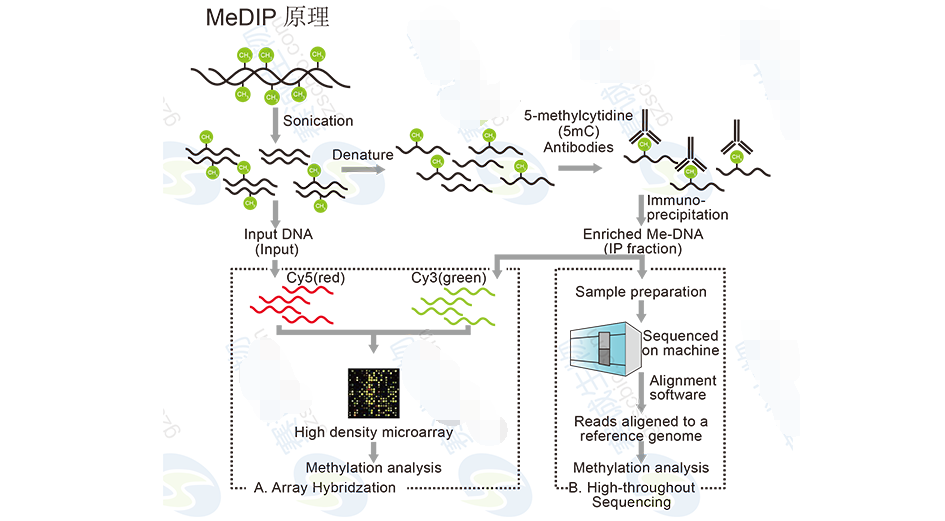
Figure 11: MeDIP principle
MeDIP-Seq Analysis Includes:1. Alignment of MeDIP-Seq reads to the reference genome.
2. Distribution of methylated regions across chromosomes and gene elements.
3. Identification of enriched regions (peaks) and their functional annotation.
4. Multi-sample comparison to detect differentially methylated regions.
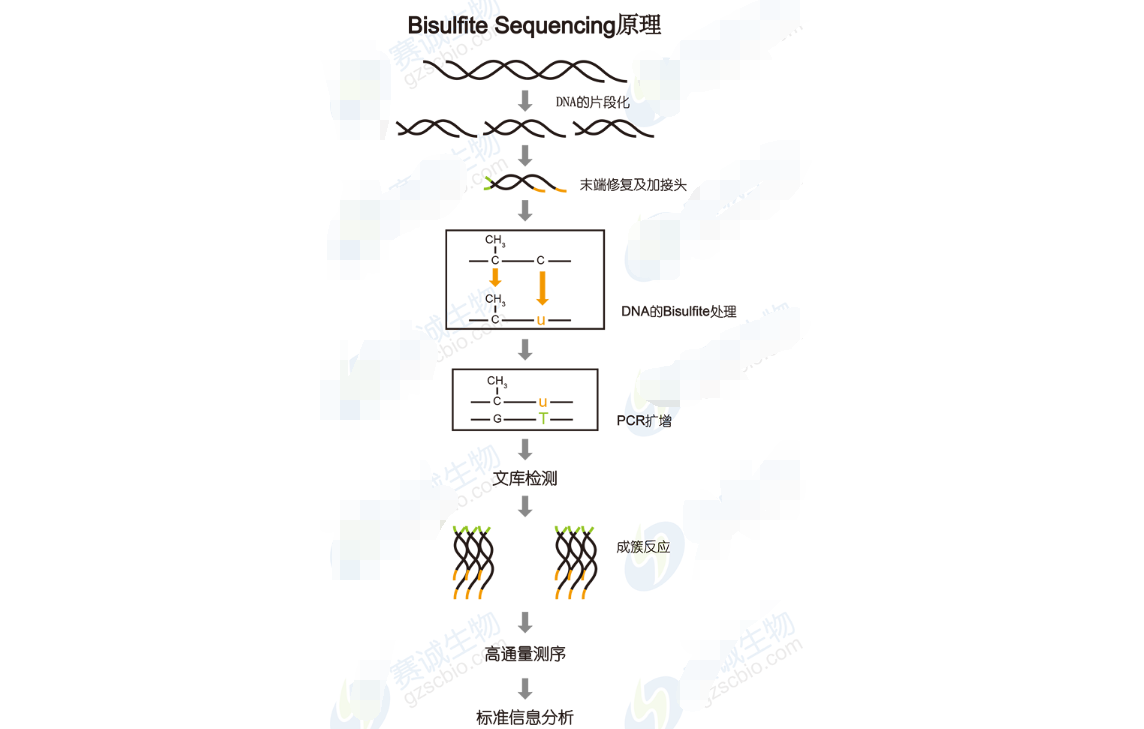
Figure 12: Bisulfite sequencing principle
Bisulfite Sequencing Analysis Includes:1. Alignment of bisulfite-treated reads to the reference genome.
2. Methylation level calculation and genome-wide distribution analysis.
3. Exploration of methylation patterns in different genomic regions and chromosome levels.
4. Detection of differentially methylated regions (DMRs) and their functional impact.
Vga Cable,Mini Data Exchange Cable,Display-Port To Vga Cable,Mini Data Exchange Adapter Wire
Dongguan City Yuanyue Electronics Co.Ltd , https://www.yyeconn.com